Published March 7, 2023

Mohit Agarwal, MD
Department of Radiology, Division of Neuroradiology, Froedtert and Medical College of Wisconsin

Karen L. Salzman, MD
Department of Radiology and Imaging Sciences, Neuroradiology Section, University of Utah

William T. O’Brien, Sr., DO
Division of Neuroradiology, Orlando Health – Arnold Palmer Children’s Hospital

Lily L. Wang, MD
Department of Radiology, University of Cincinnati College of Medicine
Deposition of protein aggregates and neuronal loss are the likely cause of cognitive decline in neurodegenerative disorders. Protein aggregates such as amyloid and tau can be imaged by amyloid and tau PET, whereas neuronal loss can be revealed by MRI and FDG PET. The pattern of protein deposition and neuronal loss may be useful in identifying the type of dementia. Cerebrovascular disease and cerebral amyloid angiopathy can cause cognitive decline on their own, or they can worsen cognitive decline caused by other neurodegenerative disorders. Normal-pressure hydrocephalus (NPH), cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) syndrome, Creutzfeldt-Jakob disease, and other conditions are additional important identifiable causes of cognitive decline on imaging.
Assessment of the mental function of an individual is done by evaluating the cognitive domains, including memory, attention, executive function, visuospatial function, language, and behavior [1]. Neurodegenerative disorders impair different aspects of these brain functions by affecting neuronal function, connectivity, and loss. Most of these diseases are characterized by accumulation of abnormal protein aggregates, which is theorized to result from dysfunction of the glymphatic system [1–3], abnormal protein processing, microglia-mediated inflammation, and dysregulated autophagy [4]. The neuronal dysfunction and subsequent neuronal loss caused by the accumulation of these protein aggregates can be identified by visual inspection or by quantification of cortical volumes or thickness, or they can be imaged by nuclear medicine techniques, such as FDG PET. The presence of certain abnormal protein deposits, such as amyloid and tau, can also be detected using PET ligands. Advanced MRI perfusion techniques such as arterial spin-labeling (ASL) can serve as a surrogate marker where hypoperfusion is indicative of underlying hypometabolism [5]. Early neuronal loss causes microstructural abnormalities in the white matter of the affected brain regions, which can be detected by quantitative diffusion MRI diffusion-tensor imaging metrics [6].
In this brief review, we will discuss the most common conditions that cause cognitive impairment and dementia.
Alzheimer Disease
Alzheimer disease (AD) is the most common form of dementia, accounting for up to 80% of the cases [1, 7, 8]. Although episodic memory and declarative memory are the most severely affected cognitive functions, patients with AD also have varying degrees of executive, language, and visuospatial function impairment [1, 7, 8]. The amyloid cascade hypothesis, which postulates that β-amyloid has a primary role in the pathophysiology, is the most widely accepted [9–12]. Deposition of tau neurofibrillary tangles, which occurs earliest in the medial temporal lobe structures, is another neuropathologic feature of AD. Tau deposition is more directly linked to neurodegeneration than is amyloid deposition, and the hippocampal and medial temporal volume loss in AD is an established structural biomarker of neuronal injury in AD [9, 12–15]. In terms of a biologic definition, amyloid biomarker positivity (Fig. 1) places an individual’s condition on the continuum of AD, but it is the positivity of both amyloid and tau markers that defines AD [9].
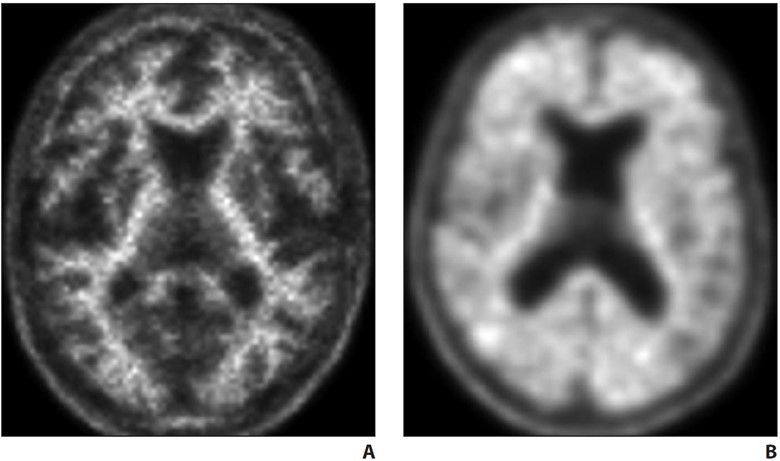
A, Negative PET examination performed with a non–glucose-based PET tracer (Amyvid, Lilly) shows no amyloid deposition in cortical regions.
B, Positive PET examination performed with same PET tracer shows extensive amyloid uptake in cortical regions. Amyloid positivity places condition on Alzheimer disease continuum.
In typical AD, there is involvement of the medial temporal lobe and lateral temporoparietal cortex, precuneus, and lateral frontal lobe, which is manifested by loss of volume and hypometabolism [16]. The Scheltens scale is widely used for visual rating of hippocampal volume loss [17] (Fig. 2).
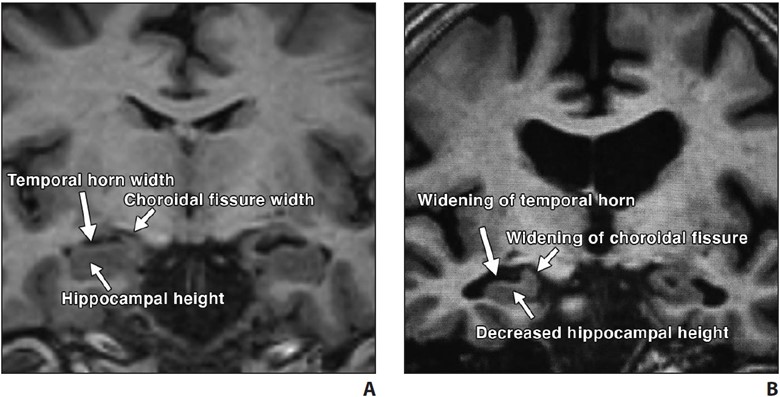
Fig. 2—Visual assessment of hippocampal atrophy performed using Scheltens scale.
A and B, Coronal T1-weighted MR images of view through hippocampus in patient with normal hippocampus (A) and in patient with mild hippocampal atrophy (B). Visual determination of hippocampal atrophy requires assessment of height of hippocampus, width of choroidal fissure, and width of temporal horn. Note near absence of CSF around hippocampus in patient with normal hippocampus (A) versus decreased hippocampal height and increased width of choroidal fissure and temporal horn in patient with mild hippocampal atrophy (B).
Hypometabolism of the involved structures is seen on FDG PET, which shows decreased activity in the lateral temporoparietal cortex, posterior cingulate cortex, precuneus, and medial temporal lobe (Fig. 3).
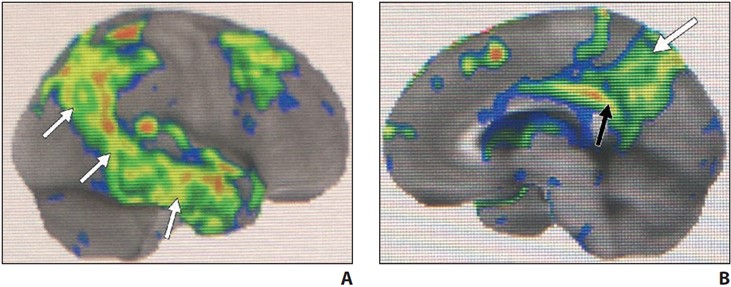
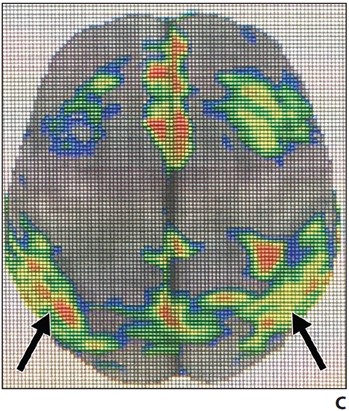
Fig. 3—FDG PET z-score maps of patient with Alzheimer disease. (Courtesy of Kleefisch C, Medical College of Wisconsin, Milwaukee, WI)
A–C, Note hypometabolism in temporoparietal (arrows, A), precuneus (white arrow, B), posterior cingulate (black arrow, B), and biparietal (arrows, C) regions.
Logopenic variant primary progressive aphasia (PPA) is the most common atypical presentation of early-onset AD, characterized by predominant language dysfunction. Impaired single-word retrieval and impaired repetition are the core features of logopenic variant PPA [18]. There is a high rate of amyloid and tau positivity [18]. Atrophy is centered at the left temporoparietal junction and the left inferior parietal and superior temporal regions, including the supramarginal and angular gyri, and is associated with concomitant hippocampal volume loss (Fig. 4).
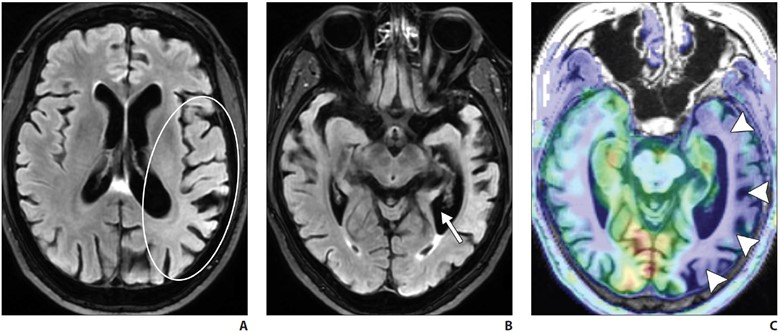
A–C, Axial FLAIR (A and B) and axial arterial spin-labeling (ASL) (C) MR images of patient with impaired word retrieval and repetition show left inferior parietal and superior temporal region atrophy, suggested by asymmetric sulcal widening on FLAIR MR image (oval, A) compared with that seen on ASL image (C). Left temporal atrophy was also suggested by asymmetric widening of left temporal horn (arrow, B). Hypoperfusion in involved regions (arrowheads, C) is likely due to underlying hypometabolism.
FDG PET shows temporoparietal hypometabolism (left greater than right) [18].
Frontotemporal Lobar Degeneration
Frontotemporal lobar degeneration (FTLD) is a heterogeneous group of disorders that typically feature progressive deterioration of behavior or language and usually are associated with neurodegeneration in the frontal and temporal lobes. FTLD is most prevalent among individuals 45–64 years old. FTLD includes six subtypes, with three predominant clinical syndromes defining these six subtypes. The most common subtype is a behavioral disorder known as behavioral variant frontotemporal degeneration. Language disorders associated with FTLD include semantic variant PPA and nonfluent or agrammatic variant PPA. Certain motor disorders included in the FTLD classification include progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and motor neuron disease (MND) [1, 7, 8].
Behavioral variant frontotemporal degeneration is the most common form of FTLD and is characterized by the gradual onset and progression of changes in personality, behavior, and executive function with relative sparing of memory and visuospatial functions (in contrast with AD). The frontal and temporal lobes are characteristically involved [7, 19, 20]. MRI shows atrophy with knife-edge gyri. On ASL and FDG PET, there is hypoperfusion and hypometabolism of the frontal and temporal lobes (Fig. 5) with relative sparing of the parietal lobes (in contrast with AD).
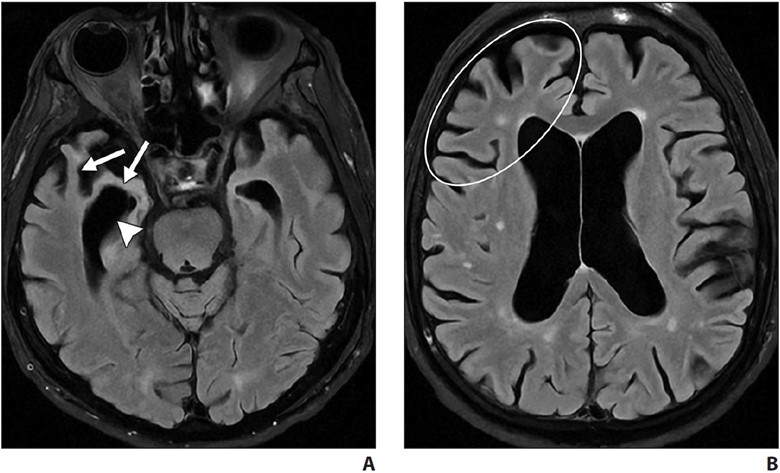
A and B, Axial FLAIR MR images of patient with personality changes show markedly asymmetric right temporal lobe and mildly asymmetric right frontal lobe atrophy. Note thinning of right temporal lobe gyri (arrows, A) and asymmetric dilatation of right temporal horn (arrowhead, A). Note asymmetric prominence of right frontal lobe sulci (oval, B) compared with left frontal lobe sulci.
Amyloid PET may also help differentiate FTD from AD, because FTD typically shows no or very little amyloid binding [7, 19–21].
Semantic variant PPA primarily presents with anomia and single-word comprehension deficits. Volume loss affects the ventral and lateral portions of the anterior temporal lobes (Fig. 6).
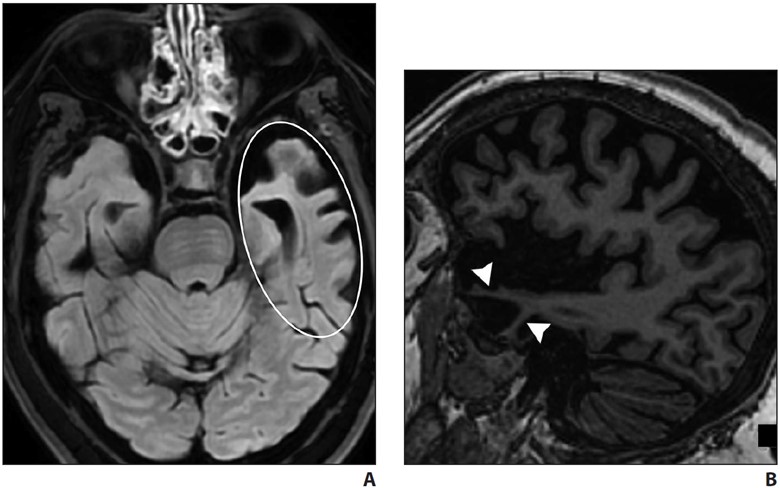
A and B, Axial FLAIR (A) and sagittal T1-weighted (B) MR images in patient with anomia and single-word comprehension deficits show asymmetric and profound anterior temporal lobe atrophy (oval, A). Note marked anterior temporal lobe gyral thinning (arrowheads, B).
In most patients, the left hemisphere is predominantly affected, although bilateral involvement may be seen. Asymmetric hippocampal volume loss is commonly seen on the side where temporal lobe atrophy is present. Tar DNA-binding protein of 43 kilodaltons (TDP-43) proteinopathy is the most common underlying neuropathology [22, 23].
Progressive nonfluent aphasia is another type of PPA on the spectrum of FTLD disorders and is associated with accumulation of tau proteins [24–27]. There is predominant involvement of the left inferior frontal region, with additional involvement of the anterior insula, prefrontal regions, supplementary motor area, and anterosuperior left temporal lobe [24–27].
Amyotrophic lateral sclerosis (ALS) is part of the FTLD-MND spectrum, with cases primarily presenting with features of either FTD or ALS. Associated FTD is more common in bulbar-onset ALS than in limb-onset ALS, with involvement of the frontal and temporal lobes. Abnormal hyperintensity is noted along the corticospinal tracts on T2-weighted and FLAIR MRI, and susceptibility-weighted imaging or gradient-recalled echo (GRE) MRI shows gyriform hypointensity along the motor cortexes [28].
PSP and CBD show mixed features of cognitive impairment and parkinsonism and an underlying tauopathy. Language symptoms, when present, are those of nonfluent or agrammatic variant PPA, which is another tauopathy. Due to the damage to the nigrostriatal dopamine pathway, 123I-FP-CIT (fluoropropyl-carbomethoxy-iodophenyl-tropane) SPECT may show abnormal reduced uptake in the basal ganglia in PSP and CBD.
Characteristic volume loss is noted in the brainstem in PSP [19, 29], manifesting as the “hummingbird” appearance on midsagittal images. Although classically described with PSP, the hummingbird sign is subjective and is commonly seen with advanced brain volume loss developing from other causes. Quantitative evaluation, with a midbrain area of less than 70 mm2 on midsagittal images or an anteroposterior diameter of the midbrain of less than 17 mm on axial T2-weighted images suggesting midbrain atrophy, could be useful [30, 31]. PSP also shows volume loss within the superior cerebellar peduncles.
CBD is rare and is characterized by unilateral or asymmetric signs of parkinsonian rigidity, myoclonus, and apraxia. Core clinical features are asymmetric progressive limb dystonia and alien limb phenomenon. The brain is atrophied asymmetrically in the perirolandic region, which is otherwise uncommon in primary neurodegenerative diseases. Other imaging features include unilateral putamen atrophy and increased hypointensity of the globus pallidi and the putamina on T2-weighted MRI and on SWI or GRE MRI. Associated unilateral cerebral peduncle atrophy may also be seen [32].
Dementia With Lewy Bodies
Dementia with Lewy bodies (DLB) is the second most common form of neurodegenerative dementia in individuals older than 65 years old. Recurrent visual hallucinations, fluctuating cognition, rapid eye movement sleep behavior disorder, and motor parkinsonism are the core clinical features. DLB can be differentiated from AD by the relative absence of medial temporal lobe atrophy [33, 34] and by greater involvement of the posterior parietal and parietooccipital regions. Atrophy within the striatal structures and brainstem is also seen in DLB. ASL and FDG PET studies show hypometabolism in the posterior parietal and occipital regions, with preservation of normal uptake in the medial temporal lobe and posterior cingulate gyrus (the cingulate island sign of DLB). Iodine- 123-labeled FP-CIT SPECT, used for imaging the dopaminergic pathway, may be useful in the workup of patients with DLB, who show decreased uptake of the tracer in the striatum, as seen in other parkinsonian diseases [33, 34].
Cerebral Amyloid Angiopathy
Deposition of amyloid-β in the media and adventitia of cerebral cortical and leptomeningeal vessels is the hallmark of cerebral amyloid angiopathy (CAA). The amyloid deposition weakens the vessel wall, leading to rupture and hemorrhage. On imaging, this manifests as a spectrum of foci of intraparenchymal lobar hemorrhage, convexal subarachnoid hemorrhage, and cortical and subcortical microhemorrhages. Areas of superficial siderosis are seen, indicative of prior subarachnoid hemorrhage. Subcortical white matter long-TR hyperintensities are typical of CAA differentiated from periventricular lesions seen in hypertensive cerebrovascular disease [35].
Acute inflammatory CAA may be seen on a background of chronic changes, in association with rapidly progressive cognitive decline [36]. On imaging, inflammatory CAA is seen as solitary or multifocal areas of confluent white matter hyperintensity with or without mass effect or patchy areas of enhancement and is often centered around foci of microhemorrhage [36] (Fig. 7).
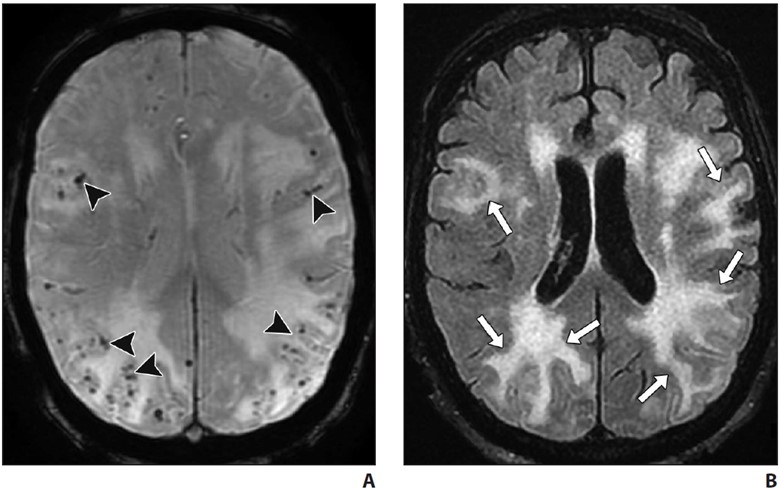
A and B, Axial susceptibility-weighted (A) and FLAIR (B) MR images in patient presenting with progressive cognitive decline. Numerous widespread foci of chronic microhemorrhage (arrowheads, A) are associated with confluent areas of vasogenic edema (arrows, B) suggesting inflammatory cerebral amyloid angiopathy. Areas of edema resolved with steroid therapy.
Normal Pressure Hydrocephalus
Primarily an idiopathic disease, NPH is characterized by progressive gait disturbance, urinary urgency or incontinence, and cognitive impairment. NPH is largely a clinical diagnosis, with imaging playing a supportive role. Objective measurements such as the Evans ratio or callosal angle have a low accuracy for diagnosis. A disproportionately enlarged subarachnoid space hydrocephalus imaging pattern, when present, has been shown to have higher accuracy where ventriculomegaly is seen along with effacement of the sulci at the vertex and multifocal enlarged sulci. Hyperdynamism may be seen on CSF flow studies, characterized by a streak of flow void through the cerebral aqueduct that is best seen on sagittal images. Radionuclide cisternography using 111In- or 99mTc-DTPA (diethylenetriaminepentaacetic acid) may show abnormal tracer reflux into the lateral ventricles and lack of tracer activity over the convexities 24–48 hours after intrathecal injection. A high volume (30–50 mL) of CSF lumbar puncture may show improved gait and cognitive testing. Improvement of gait disturbance after lumbar puncture has a high PPV for favorable postintervention results [37].
Creutzfeldt-Jakob Disease
A fatal prion disease, CJD presents with rapidly progressive dementia, along with myoclonus, pyramidal, extrapyramidal, and cerebellar signs. CJD is mostly sporadic, but it can be familial, infectious (variant CJD), or iatrogenic. There is spongiform degeneration and gliosis. The MRI sequence with the highest sensitivity and specificity is DWI, which shows the characteristic imaging finding of hyperintensity of the basal ganglia, thalami, and the cortical regions [38, 39] (Fig. 8).
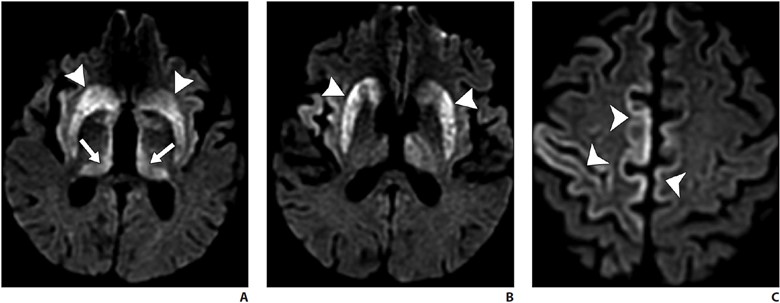
A–C, Axial diffusion-weighted MR images of patient presenting with rapidly progressive cognitive decline show signal abnormality in bilateral corpus striatum (arrowheads, A and B), thalamic pulvinar (arrows, A) and scattered cortical gyriform reduced diffusion (arrowheads, C). Hockey-stick configuration of dorsomedial and pulvinar involvement is commonly described with Creutzfeldt-Jakob disease.
Symmetric involvement of the posterior thalami (pulvinar sign) and the dorsomedial nuclei (hockey-stick sign) is common in variant CJD but can also be seen in sporadic CJD. Bilateral but asymmetric involvement of the cortical areas is seen [38, 39]. Enhancement is uncommon. Definitive diagnosis may require brain biopsy. Death ensues in a few months.
Vascular Contributions to Cognitive Impairment and Dementia
Cerebrovascular small-vessel disease is a common complication of uncontrolled hypertension, which frequently affects the periventricular white matter seen as white matter hyperintensities on T2 FLAIR imaging. Hypertensive arteriopathy, amyloid angiopathy, or genetic causes of cerebrovascular disease (such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy [CADASIL]) can cause cognitive impairment by interrupting brain networks traversing the affected white matter regions [40, 41]. The likelihood of vascular disease contributing to cognitive dysfunction increases with increasing small-vessel disease and infarct burden.
CADASIL is the most common genetic cause of adult-onset cerebrovascular disease. Migraine with aura, stroke, and chronic presentations, such as cognitive decline and dementia, are the clinical features. Patients most frequently present in the 3rd decade of life, and most patients present by late middle age. When patients present earlier than the 3rd decade, subcortical FLAIR white matter hyperintensities are seen, which over the years progress to the characteristic confluent hyperintensities in the anterior temporal poles, superior frontal lobes, and the external capsules [42].
Deposition of protein aggregates and neuronal loss are the likely cause of cognitive decline in neurodegenerative disorders. Protein aggregates such as amyloid and tau can be imaged by amyloid and tau PET, whereas neuronal loss can be shown on MRI and FDG PET. The pattern of protein deposition and neuronal loss may be useful in identifying the type of dementia. Cerebrovascular disease and cerebral amyloid angiopathy can cause cognitive decline on their own or worsen cognitive decline due to other neurodegenerative disorders. NPH, CADASIL, CJD, and other conditions are additional important identifiable causes of cognitive decline on imaging.
We thank Christopher Kleefisch of the Medical College of Wisconsin for his contribution to the PET images.
References
- Gliebus GP. Memory dysfunction. Continuum (Minneap Minn) 2018; 24:727–744
- Plog BA, Nedergaard M. The glymphatic system in central nervous system health and disease: past, present, and future. Annu Rev Pathol 2018; 13:379– 394
- Zhang L, Chopp M, Jiang Q, Zhang Z. Role of the glymphatic system in ageing and diabetes mellitus impaired cognitive function. Stroke Vasc Neurol 2019; 4:90–92
- Guo F, Liu X, Cai H, Le W. Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol 2018; 28:3–13
- Alsop DC, Detre JA, Golay X, et al. Recommended implementation of arterial spin-labeled perfusion MRI for clinical applications: a consensus of the ISMRM perfusion study group and the European consortium for ASL in dementia. Magn Reson Med 2015; 73:102–116
- Shim G, Choi KY, Kim D, et al. Predicting neurocognitive function with hippocampal volumes and DTI metrics in patients with Alzheimer’s dementia and mild cognitive impairment. Brain Behav 2017; 7:e00766
- Bhogal P, et al. The common dementias: a pictorial review. Eur Radiol 2013; 23:3405–3417
- Tartaglia MC, Vitali P, Migliaccio R, Agosta F, Rosen H. Neuroimaging in Dementia. Continuum (Minneap Minn) 2010; 16:153–175
- Jack CR Jr, Bennett DA, Blennow K, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018; 14:535–562
- Dolci GAM, Damanti S, Scortihini V, et al. Alzheimer’s disease diagnosis: discrepancy between clinical, neuroimaging, and cerebrospinal fluid biomarkers criteria in an Italian cohort of geriatric outpatients: a retrospective cross-sectional study. Front Med (Lausanne) 2017; 4:203
- McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging–Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7:263–269
- Jack CR Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013; 12:207–216
- Burnham SC, Coloma PM, Li QX, et al. Application of the NIA-AA research framework: towards a biological definition of Alzheimer’s disease using cerebrospinal fluid biomarkers in the AIBL study. J Prev Alzheimers Dis 2019; 6:248–255
- Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging–Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7:280–292
- Fox NC, Schott JM. Imaging cerebral atrophy: normal ageing to Alzheimer’s disease. Lancet 2004; 363:392–394
- Ferreira D, Verhagen C, Hernández-Cabrera JA, et al. Distinct subtypes of Alzheimer’s disease based on patterns of brain atrophy: longitudinal trajectories and clinical applications. Sci Rep 2017; 7:46263
- Scheltens P, Leys D, Barkhof F, et al. Atrophy of medial temporal lobes on MRI in “probable” Alzheimer’s disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol Neurosurg Psychiatry 1992; 55:967–972
- Rohrer JD, Rossor MN, Warren JD. Alzheimer’s pathology in primary progressive aphasia. Neurobiol Aging 2012; 33:744–752
- Agosta F, Galantucci S, Magnani G, et al. MRI signatures of the frontotemporal lobar degeneration continuum. Hum Brain Mapp 2015; 36:2602–2614
- Irwin DJ, Cairns NJ, Grossman M, et al. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol 2015; 129:469–491
- Meijboom R, Steketee RME, de Koning I, et al. Functional connectivity and microstructural white matter changes in phenocopy frontotemporal dementia. Eur Radiol 2017; 27:1352–1360
- Chen Y, Chen K, Ding J, et al. Brain network for the core deficits of semantic dementia: a neural network connectivity–behavior mapping study. Front Hum Neurosci 2017; 11:267
- Joubert S, Vallet GT, Montembeault M, et al. Comprehension of concrete and abstract words in semantic variant primary progressive aphasia and Alzheimer’s disease: a behavioral and neuroimaging study. Brain Lang 2017; 170:93–102
- Gorno-Tempini ML, Miller BL. Primary progressive aphasia as a model to study the neurobiology of language. Brain Lang 2013; 127:105
- Gorno-Tempini ML, Brambati SM, Ginex V, et al. The logopenic/phonological variant of primary progressive aphasia. Neurology 2008; 71:1227–1234
- Gorno-Tempini ML, Dronkers NF, Rankin KP, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol 2004; 55:335– 346
- Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011; 76:1006–1014
- Conte G, Sbaraini S, Morelli C, et al. A susceptibility-weighted imaging qualitative score of the motor cortex may be a useful tool for distinguishing clinical phenotypes in amyotrophic lateral sclerosis. Eur Radiol 2021; 31:9
- Rohrer JD, Warren JD. Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol 2011; 24:542–549
- Warmuth-Metz M, Naumann M, Csoti I, Solymosi L. Measurement of the midbrain diameter on routine magnetic resonance imaging: a simple and accurate method of differentiating between Parkinson disease and progressive supranuclear palsy. Arch Neurol 2001; 58:1076–1079
- Oba H, Yagishita A, Terada H, et al. New and reliable MRI diagnosis for supranuclear palsy. Neurology 2005; 64:2050–2055
- Saeed U, Compagnone J, Aviv RI, et al. Imaging biomarkers in Parkinson’s disease and Parkinsonian syndromes: current and emerging concepts. Transl Neurodegener 2017; 6:8
- Watson R, O’Brien JT, Barber R, Blamire AM. Patterns of gray matter atrophy in dementia with Lewy bodies: a voxel-based morphometry study. Int Psychogeriatr 2012; 24:532–543
- Watson R, Colloby SJ, Blamire AM, O’Brien JT. Subcortical volume changes in dementia with Lewy bodies and Alzheimer’s disease: a comparison with healthy aging. Int Psychogeriatr 2016; 28:529–536
- Wardlaw JM, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013; 12:822–838
- Salvarani C, et al. Imaging findings of cerebral amyloid angiopathy, Aβ-related angiitis (ABRA), and cerebral amyloid angiopathy-related inflammation: a single-institution 25-year experience. Medicine (Baltimore) 2016; 95:e3613
- Graff-Radford NR. Normal pressure hydrocephalus. Neurol Clin 2007; 25:809–832
- Kallenberg K, Schulz-Schaeffer WJ, Jastrow U, et al. Creutzfeldt-Jakob disease: comparative analysis of MR imaging sequences. AJNR 2006; 27:1459–1462
- Tian HJ, Zhang JT, Lang SY, Wang XQ. MRI sequence findings in sporadic Creutzfeldt-Jakob disease. J Clin Neurosci 2010; 17:1378–1380
- Koga H, Takashima Y, Murakawa R, Uchino A, Yuzuriha T, Yao H. Cognitive consequences of multiple lacunes and leukoaraiosis as vascular cognitive impairment in community-dwelling elderly individuals. J Stroke Cerebrovasc Dis 18:32–37
- Sweeney MD, Montagne A, Sagare AP, et al. Vascular dysfunction: the disregarded partner of Alzheimer’s disease. Alzheimers Dement 2019; 15:158–167
- Ferrante EA, Cudrici CD, Boehm M. CADASIL: new advances in basic science and clinical perspectives. Curr Opin Hematol 2019; 26:193–198